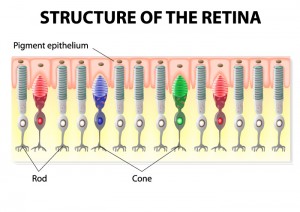
No, people with inherited retinal diseases don’t have to adopt new names or personas, or go into witness protection programs, to save their vision. But by changing the identity of cells in the retina — namely rods — researchers may someday be able to slow or halt vision loss for those with retinitis pigmentosa (RP) and other related conditions.
While the innovative therapeutic approach is not ready to be tested in humans, a research team led by Tom Reh, PhD, University of Washington, and Sheng Ding, PhD, University of California, San Francisco, accomplished the feat in mice with RP. The investigators treated rods in the mice with a compound known as photoregulin1 (PR1) that blocked a gene involved in rod development called Nr2e3. That, in turn, reduced the expression (activity) of other rod-associated genes, making the rods less rod-like and more like cones. Doing so stopped retinal degeneration, preserving both rods and cones. Rods and cones are important, because they’re the cells that make vision possible. Results of the PR1 study were published online in the journal Investigative Ophthalmology & Visual Science.
Drs. Reh and Ding, along with their colleagues Paul Nakamura, PhD, and Shibing Tang, PhD, targeted rods because they’re the photoreceptors that degenerate first in RP and some other retinal diseases. Most gene mutations that cause RP affect rod function and health. Because cones depend on rods for survival, cones die off as rods are lost. Therefore, saving rods should save cones.
There is a downside to PR1 therapy: Though the converted rods survive, they no longer process light to provide rod-mediated vision. However, the converted rods preserve existing cones. By saving cones, the therapy would maintain the patient’s central and color vision and vision in bright settings, as well as the ability to perceive details (i.e., read, drive, and recognize faces). With the loss of rod-mediated vision, the therapy recipient’s night and peripheral vision would be significantly reduced.
Dr. Reh notes that much more work in chemistry needs to be done to develop PR1 into an oral compound that could be taken by people in a clinical trial.
“I think this approach has the potential to help anyone who still has rods left in their retinas,” says Dr. Reh. “We were pleased that PR1 worked in mice with a mutation in RHO, which causes autosomal dominant RP, and a mutation in PDE6β, which causes autosomal recessive RP. We would like to test the compound in a variety of retinal-disease models to see how broad the efficacy is.”