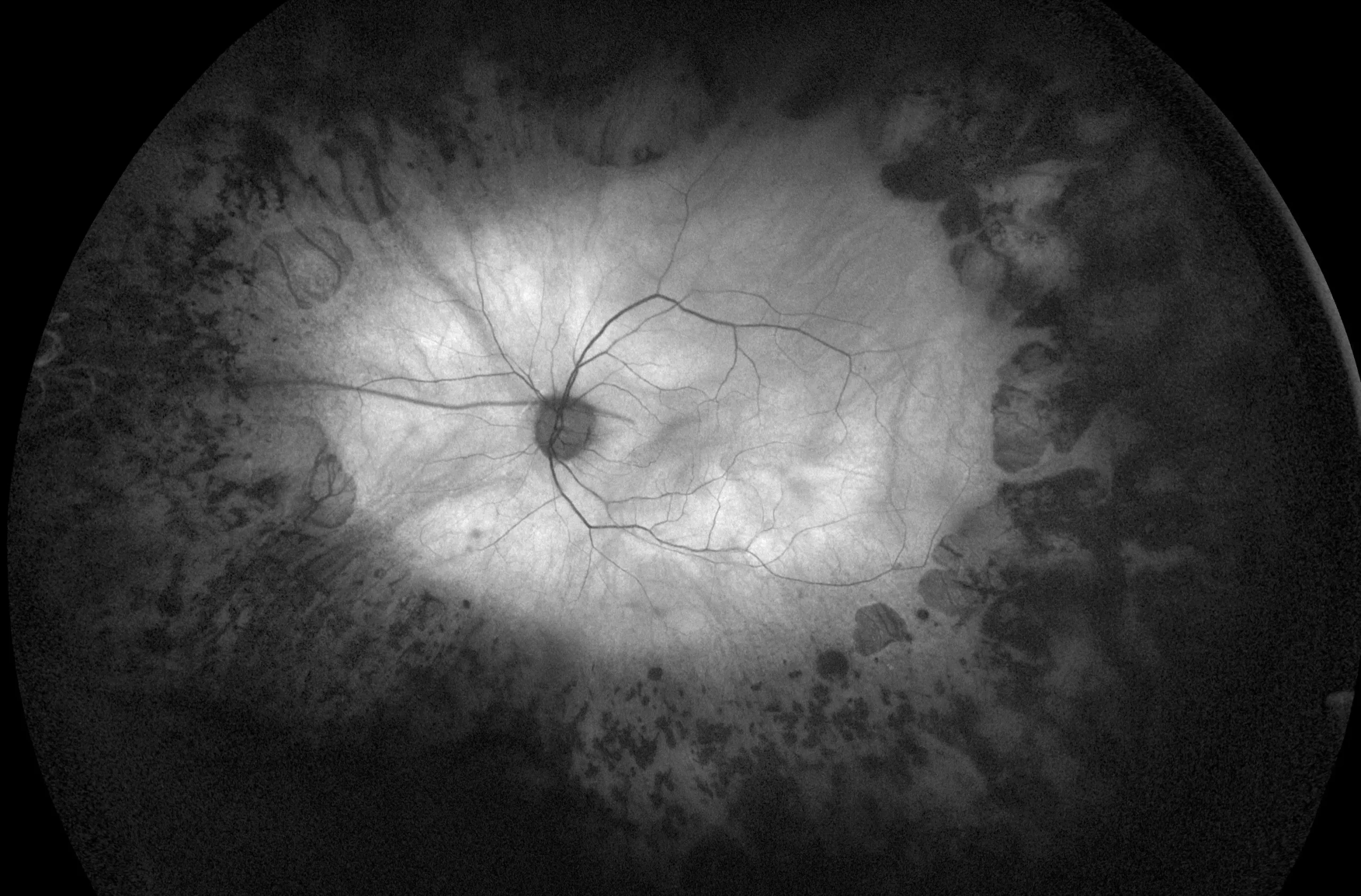
در این مطالعه عوامل ژنتیکی دخیل در سندرم باردت بیدل در 18 بیمار ایرانی مورد بررسی قرار گرفت. در مرحله اول، با توجه به نتایج WES، ما توانستیم هشت نوع، به طور کلی، چهار جهش جدید از جمله جهش بی معنی در BBS6 (c.1334T > G)، حذف جزئی (c.602 -12_622del) در BBS7 را شناسایی کنیم. حذف فریم شیفت (c.131-143del) در BBS17، و جهش بی معنی (c.901-905del) در BBS8، و چهار جهش شناخته شده مانند، جهش بی معنی
زودرس (c.C1780T) در BBS2 در دو بیمار، جهش غیرمعنا ( c.790G > A) در BBS5، جهش frameshift (c.271dup) در BBS10، و جهش بی معنی (1063C> T) در BBS12 شناسایی شد.
در مرحله بعد، با تجزیه و تحلیل مجدد موارد حل نشده، یک جهش مترادف (c.471G > A) را در هفت بیمار شناسایی کردیم. شش بیمار از قومیت بلوچ و یک بیمار از قومیت فارس بودند. این مطالعه جداسازی این جهش را تایید کرد. ارزیابی نواحی هموزیگوت در شش بیمار، وجود ناحیه هموزیگوتی مشترک را در چهار بیمار از قومیت بلوچ نشان داد که احتمال ماهیت پایه این جهش را نشان می دهد. این رویکرد با این احتمال توجیه میشود که والدین بیتأثیر که فامیل دور هستند، از یک گروه قومی با نرخ درونهمسری بالا آمدهاند، یا در یک ایزوله جغرافیایی زندگی میکنند، میتوانند هتروزیگوتها برای همان جهش مغلوب از یک اجداد مشترک باشند.
این جهش یک جهش مترادف است که در نگاه اول یک جهش خوش خیم به نظر می رسد، اما مطالعات جمعیتی گذشته نشان داده است که این جهش با بیماری BBS مرتبط است [16]. با توجه به بالا حفاظت از آخرین نوکلئوتید گوانین و نقش احتمالی آن در پیرایش، این جایگزینی هموزیگوت در BBS2، c.G471A (p.T157T) در نظر گرفته میشود که تأثیر قوی بر فرآیند پیرایش قبل از mRNA دارد. تجزیه و تحلیل HSF سایت اهداکننده نوع وحشی را به دلیل این جهش شناسایی کرد. اختلال در فرآیند پیرایش احتمالا منجر به حفظ اینترون در mRNA بالغ می شود و با ایجاد جهش تغییر فریم و کدون خاتمه زودرس، احتمالاً منجر به NMD می شود. بنابراین، تعامل پروتئین BBS2 با سایر پروتئینهای موجود در مجتمع BBSome ، مجموعهای از پروتئینهای دخیل در رشد اولیه مژههای سلولی، مختل میشود.
اطلاعات دقیقی در مورد فراوانی این جهش در پایگاه های داده ایرانی وجود ندارد و فراوانی آللی این جهش در پایگاه داده GnomAD 0.000014 گزارش شده است [19]. بر اساس موقعیت جغرافیایی بیماران مبتلا به این جهش در این مطالعه و مطالعات قبلی، ظاهراً این جهش در این منطقه بسیار قدیمی است. این امر احتمال فراوانی بالای این جهش را افزایش می دهد
جامعه ایرانی بنابراین غربالگری جمعیتی این جهش در جامعه جنوب شرق ایران می تواند تاثیر خوبی در پیشگیری از عود این بیماری داشته باشد.
در دهه گذشته، تغییرات تعداد کپی (CNVs) به عنوان عامل اصلی بار ژنتیکی در اختلالات نادر و شایع شناخته شده است. CNV یک پدیده ناشی از بازآرایی ژنومی است و طول آن معمولاً بیش از 1 کیلوبایت است [20]. این تغییرات نقش بسزایی در ایجاد تنوع لازم در جمعیت و فنوتیپ بیماری دارد. در این مطالعه، بررسی CNV در یک بیمار منجر به شناسایی یک حذف بزرگ در ژن BBS1 که شامل اگزون های 14 تا 17 می باشد، شد. این حذف با استفاده از روش Quantitative real-time PCR در اعضای خانواده بیماران تایید شد. حذف این ناحیه منجر به حذف دامنه های انتهایی و ایجاد پروتئین کوتاه شده و بی اثر می شود.
علاوه بر این، به دلیل ماهیت ناهمگن BBS، توزیع ژن های عامل در بین جمعیت ها متفاوت است. اگرچه بیشترین نسبت ژن های درگیر در BBS در جمعیت های مختلف متعلق به BBS1 و BBS10 است [21]، در این مطالعه 50 درصد از بیماران ایرانی الاصل حامل جهش BBS2 بودند.
به دلیل پدیده جهش بنیانگذار BBS2. این اطلاعات کاملاً با جمعیت های قفقازی متفاوت است. این نتایج رویکرد جدیدی را برای تشخیص BBS با استفاده از ژنتیک مولکولی در ایران و شاید دیگر کشورهای خاورمیانه به ویژه پاکستان و افغانستان پیشنهاد میکند.
یافتن جهشهای مکرر یا پایهگذار مخصوص جمعیت میتواند ابزاری حیاتی برای توسعه یک الگوریتم کارآمد برای عملکرد ژنتیکی بالینی باشد که پروتکلهای غربالگری را قبل از استفاده از فناوریهای پیشرفتهتر مانند توالییابی نسل بعدی اصلاح میکند.
نتیجه گیری : بیماری های BBS مهم ترین عضو ایلیوپاتی است که منجر به عقب ماندگی ذهنی سندرمی همراه با رتینوپاتی، پلی داکتیلی، چاقی، اختلالات تولید مثلی و کلیوی می شود. تاکنون تعداد زیادی از ژنهای دخیل در سیلیوژنز در ارتباط با BBS شناسایی شدهاند. با این حال، بخش قابل توجهی از این بیماری ایدیوپاتیک باقی می ماند. علاوه بر شناسایی جهش های جدید مرتبط با این بیماری، مطالعات بیشتری برای شناسایی انواع غیرعادی در ژن های شناخته شده مورد نیاز است. دادههای ما در این مطالعه نشان میدهد که نوع c.G471A یک جهش پایهگذار است که باعث ایجاد Barde میشود.
منبع مقاله : www.researchsquare.com